| distilled water | 4.4 |
| anchored oligo dT primers * | 3.0 |
| 10 x Taq buffer | 1 |
| Taq polymerase | 0.1 |
| 10 x DIG-dUTP/dNTP mix ** | 1.5 |
| cDNA insert (> 3.5 ng/ | 0.6 |
- * anchored oligo dT primers
- 5.3
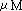 (dT)17dG / 5.3 (dT)17dC
/ 21.3 (dT)17dA
(dT)17dG / 5.3 (dT)17dC
/ 21.3 (dT)17dA
(This is to avoid the effect of poly-A stretch. Other vector primers may be used.) - ** 10 x DIG-dUTP/dNTP mix
- 0.35mM DIG-dUTP / 0.65mM dTTP / 1mM each d(A, G, C)TP
| hot start at 95oC for 45 sec | |
| 95oC x 15 sec | |
| 45oC x 1 min | |
| 72oC x 1 min |